Expert Q&A: Can You Prevent Alzheimer’s?

Why do some people get Alzheimer's, while others are protected? Harvard Alzheimer's researcher Dr. Rudy Tanzi offers insights on the neurodegenerative process of Alzheimer's, and why some people are more resilience against the disease than others.
While you might read reports about the ongoing debate over what causes Alzheimer’s, Dr. Rudy Tanzi, an Alzheimer’s researcher at Massachusetts General Hospital in Boston and professor of neurology at Harvard University, says that scientists settled that debate several years ago. According to Tanzi, most scientists believe there is no question that beta-amyloid, the protein that forms into plaques in Alzheimer’s patients’ brains, leads to Alzheimer’s. He says that anyone who is still confused about the beta-amyloid hypothesis should look at his team’s 2014 study that illustrates how beta-amyloid causes plaques, tangles and eventually, inflammation in the brain that results in dementia.
While Dr. Tanzi says the debate over the beta-amyloid hypothesis has been put to rest, some still question why there are people with plaques and tangles in their brain who don’t develop the symptoms of Alzheimer’s: devastating loss of function and memory. Others wonder why an anti-inflammatory drug like Ibuprofen can’t alleviate Alzheimer’s symptoms, if researchers believe that neuroinflammation causes Alzheimer’s.
Being Patient spoke to Dr. Tanzi about these questions, his research on what happens in an Alzheimer’s patient’s brain, how the herpes virus triggers Alzheimer’s symptoms and promising clinical trials for drugs that could tackle beta-amyloid. He discussed why previous clinical trials have failed and where future studies are headed. This is part one of a two-part series on Being Patient’s discussion with Dr. Tanzi on Alzheimer’s research and prevention.
Being Patient: You’ve been in this field for over 35 years. Since we spoke last year, what has been the biggest advancement in Alzheimer’s research?
Dr. Tanzi: Over 100 years ago, Dr. Alois Alzheimer first described the plaques and tangles that he believed were causing dementia. He was examining a female with early-onset Alzheimer’s. However, for decades, we didn’t know how these plaques and tangles worked or were involved in the disease. In the ’80s and ’90s, when we discovered the first Alzheimer’s genes, including the amyloid gene, which I named amyloid precursor protein (APP), amyloid became the biggest drug target in the industry for Alzheimer’s. We still didn’t know if amyloid caused the disease, because we put these genes in mice; the mice had some amyloid and eventually developed issues, but they didn’t get the tangles. There was a disconnect.
It wasn’t until Doo Yeon Kim in our group invented what we call ‘Alzheimer’s in a dish’ that we knew amyloid causes Alzheimer’s. We took mini human brain organoids [clumps of brain cells the researchers used to develop ‘mini-brains’], grew them in a petri dish, put the Alzheimer’s genes in them and then looked at when they made plaques. After putting the Alzheimer’s genes that we discovered in the human brain organoids, we saw tangles, and when we stopped the plaques, we stopped the tangles. That was the first real proof of concept—only several years ago. When studying the human-brain organoid rather than mice, we found that plaques cause tangles. From there, you get what’s called neuroinflammation, and then eventually, dementia.
Being Patient: There’s been a lot of news about viruses recently, especially the herpes virus. You were behind that research. What is the connection between the herpes virus and Alzheimer’s disease?
Dr. Tanzi: Let’s start with why we make amyloid. We now know that amyloid accumulates in the brain 10 or 15 years before Alzheimer’s symptoms appear. A lot of these clinical trials that targeted amyloid failed because they were done too late. Amyloid is the match and the tangles are the brush fires. The brain’s immune system then reacts to the tangles that develop, and little pockets of dying nerve cells, with inflammation; neuroinflammation is the brain’s response to plaques, tangles and pockets of cell death. Neuroinflammation then kills 10 or 100 times more nerve cells, and then you develop dementia.
Amyloid leads to the plaques, tangles are the brush fires and neuroinflammation is the forest fire. The clinical trials that failed hit amyloid too late. They tried to put out a forest fire without blowing out the match first. Now, people are doing trials that hit people very early, even before symptoms occur, to stop the amyloid.
Why do we get amyloid? Who strikes the match? In our lab, we found that amyloid forms around viruses like herpes viruses—or bacteria and fungus, like yeast—instantly. Within 24 hours, you get a plaque, with the virus trapped inside of it. These are called extracellular traps; they’re part of our innate immune system. Antibodies take a while to kick in when we get an infection, so before that, our primitive immune system tries to help us. Plaques in the brain are actually extracellular traps—part of the brain’s immune system that tries to trap viruses like herpes.
We’re not saying you have to have a virus or bacteria to make a plaque; there’s other ways to make a plaque, like through genetics. We’re also not saying that viruses like herpes cause Alzheimer’s. However, we found that viruses—namely, HHV-6, HHV-7 and to a lesser extent, the herpes simplex virus 1 that causes cold sores—reactivate in old age. When they reactivate, the amyloid instantly gets seated, forms a big mass around the virus and traps it to protect the nerve cells in the brain. We think that’s what’s striking the match and causing amyloid.
Of course, we know from the familial early-onset genes I discovered decades ago that certain genetic factors may cause those plaques anyway, even without a virus or bacteria.
Being Patient: Is the virus a trigger to this process, which gives us an unhealthy level of beta-amyloid?
Dr. Tanzi: Yes. We think that you need a small amount of amyloid-beta protein to protect the brain. The brain was once thought to be sterile, but viruses and bacteria get reactivated, especially as we get older. The immune system and the blood-brain barrier (BBB) that keeps out pathogens weaken as you age. When we develop an infection, this amyloid-beta protein binds to the microbe causing the disease, traps and neutralizes it. We think this process is one of the triggers for Alzheimer’s that leads to the plaques, tangles and neuroinflammation.
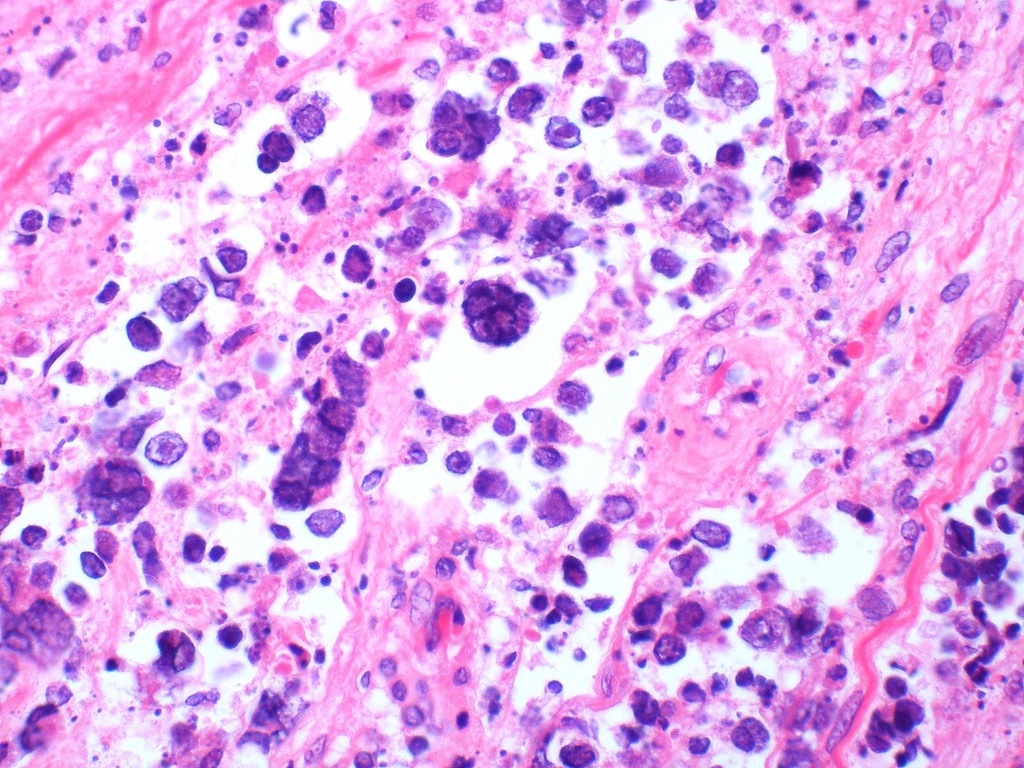
Tanzi and his team found that herpes viruses, including herpes simplex 1, cause processes in the brain that may lead to Alzheimer’s. | © Yale Rosen / Flickr
Being Patient: Why do some people live with plaques, but don’t develop Alzheimer’s?
Dr. Tanzi: Those are called resilient brains. Occasionally, someone passes at 80 or 90 who never had dementia. In these resilient brains, you see levels of plaques and tangles that make you say, “This should’ve been an Alzheimer’s patient, so why weren’t they?” Somehow, their nerve cells didn’t die because they didn’t have neuroinflammation. The brain’s immune system stayed “chilled out” and did not react to those plaques, tangles and pockets of ensuing cell death with an inflammatory response. Because of that, it didn’t kill the 10 or 100 times more nerve cells that plaques and tangles kill, so these individuals did not develop dementia. That’s good news because it means that technically, we can live with a lot of plaques and tangles as long as the brain’s immune system doesn’t react against them with neuroinflammation.
With neuroinflammation, glial cells—the brain’s housekeepers—get worked up and say, “We have to protect the brain. Nerve cells are dying. There must be an attack or infection.” These cells—the scrubbing bubbles that clean the brain and plaque (especially at night when we sleep)—become soldiers that shoot off free radicals that kill nerve cells. Even though they’re just trying to clean off the area with friendly fire, this collateral damage kills more nerve cells than the plaques and tangles. Some people don’t have this immune response to the plaques and tangles, so they’re protected from Alzheimer’s, despite the presence of the plaques and tangles.
Being Patient: Is that why some researchers have become skeptical of the beta-amyloid hypothesis and how plaques are related? If people have a lot of plaques and tangles in their brain, but don’t develop Alzheimer’s, could it be possible that researchers are looking in the wrong place?
Dr. Tanzi: That interpretation was debated for decades because in mice, we put in these mutations that guaranteed early-onset Alzheimer’s by 60 years old—these terrible mutations, including the amyloid-precursor protein gene and the presenilin genes that were discovered in 1995. These genes all have one thing in common: They lead to more amyloid in the brain. However, when you put these genes in mice, they got amyloid, but they didn’t get the tangles or develop Alzheimer’s.
However, now we can put this debate to rest. If people are still skeptical about whether beta-amyloid leads to Alzheimer’s, they can read our 2014 study in Nature. We found that in human brain organoids, if you have the plaques, tangles follow, and if you stop the plaques, you stop the tangles. We know now that plaques come very early. I think most people who really take the time to read the scientific literature will not argue with the amyloid hypothesis. The ones who just read about controversy over this theory in a local newspaper might argue about it, but most scientists have come to an agreement. To get the disease, plaques and tangles have to cause neuroinflammation, or you won’t get symptoms. That’s why there are these rare cases with plaques and tangles, but no dementia; inflammation didn’t kick in.
We’ve also found genetic factors lead to neuroinflammation. We found the first Alzheimer’s gene that causes neuroinflammation—CD33—and another, TREM2. Those genes control the neuroinflammation in the plaques and tangles. Some people have protective mutations in those genes, so despite the plaques and tangles, they don’t get neuroinflammation and remain symptom free. I think we can explain all of this now without controversy.
Being Patient: If researchers think that if you don’t get inflammation, you won’t get Alzheimer’s, why wouldn’t people just take an anti-inflammatory for the rest of their lives?
Dr. Tanzi: Anti-inflammatories like Ibuprofen don’t work very well in the brain or don’t get into the brain. Also, neuroinflammation in the brain is very different from inflammation you get when you cut your knee. There’s different types of cells involved. Microglial cells and astrocytes are the brain’s worker bees or housekeepers. While they are normally nurturing, when they think something’s wrong or smell nerve cells dying—even just a few here and there from plaques and tangles—they kick in and say, “Wipe out the area.” That’s neuroinflammation. To quell that, you can’t just use a simple steroid or anti-inflammatory like Ibuprofen. Since we discovered the genes that control neuroinflammation, we’re now doing screens. We’ve actually found drugs that control that neuroinflammatory process. One of them is even in a Phase 3 clinical trial right now and we have great hope for it. It’s a repurposed drug that’s been reformulated by a company I’ve been involved with. So far, things are looking optimistic, but we’ll see what happens.

According to Tanzi, taking an anti-inflammatory drug like Ibuprofen doesn’t work as well in the brain. | © Vnukko / Pixabay
Being Patient: How long before we will know the results of the trial and how effective that drug could be?
Dr. Tanzi: We won’t know the results for another year or so. It might even go up to 2020. Sometimes, companies can do what I call interim analyses. Even though the data is large, you can look and see if some of the patients are getting worse and some are getting better, since we have a placebo. You always want to look for separation and to see patients going in different directions. That gives you hope that there might be something going on, even if you don’t know who’s on the drug or who’s a placebo. There are many things you can do along the way to take a sneak peak and see if things are going well, but that’s up to them.
This interview has been edited for length and clarity.
Related Articles
No Related Post Found





Thank you very much for the valuable and clear information. Many will benefit. My question: can I quote from this interview? And if yes what would be the appropriate vote for quotation. I assuming it is not copyright protected because we have the option to share it with social media?
Hi Marion,
We don’t mind if you quote as long as you attribute to Being Patient and link back to our site http://www.beingpatient.com. Thank you for asking!
Does Dr. Tanzi and Snow take the supplements themselves or does anyone in their family just in case. Do they have the ApoE4? How is it possible that nobody asked them if their supplements are in any human clinical trials, and if not, why? If it is ready to be sold to people hoping to prevent ALZ 20 years from now, $50 a month times 12 x 20 = A VERY LARGE INVESTMENT IN SOMETHING THAT MIGHT PREVENT ALZ? But one wouldn’t just stop at any age because they could still get tangles so basically I’m at 47 years old looking to spend $50 a month just on one of the supplements for the rest of my life? I figure once I add in Niagen, Ashwaganda and the 6 others I’ve found, I could be looking at $200 a month for the rest of my life and I didn’t have an income right now.
What is the name of the company in phase 3 trial??
Very interesting article Have been having problems with memory since teenage Can never understand why my brain sblocks out a lot of things
Please keep me informed.
My late Mother had Dementia and I am terrified that I may get it.
I had two brain aneurisms sixteen years ago which were clipped and I am pretty well over it all.
I am very prone to fever blisters on my nose, but that is due to a post nasal drip.
I also have a yeast problem.