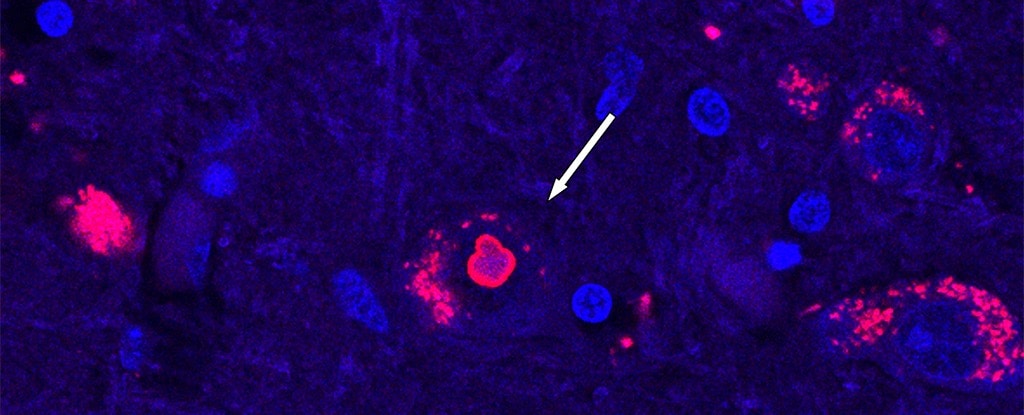
Scientists say they've pinpointed the exact gene fragment that causes a rare hereditary form of a degenerative neurological disorder called spinocerebellar ataxia, giving families affected by it hope for targeted treatments and earlier detection.
In the 1840s, near Utah’s Great Salt Lake, a little piece of DNA went haywire. This genetic malfunction gave rise to a rare disorder that caused those affected to lose control over their balance and muscle movement. The condition was degenerative, and incurable. And it has been passed down, generation to generation ever since. If scientists can figure out exactly what stretch of genetic code drives a disorder like this one, it could open the door to better diagnostics, targeted treatments, even a cure. Scientists have been working for some 25 years to find the cause of this disorder — spinocerebellar ataxia type 4 — and now, an international team thinks they’ve found it.
According to new research published in the journal Nature Genetics, an international group of scientists, including researchers at University of Utah, say they’ve cracked the code of spinocerebellar ataxia type 4, a group of progressive disorders that affect the cerebellum — the part of the brain that controls movement.
Approximately 150,000 Americans live with a form of SCA. The symptoms of Type 4 include balance issues, muscle weakness (atrophy), and difficulty coordinating body movements (known as ataxia), which causes jerky, unsteady gait and difficulty speaking (known as dysarthria). And thanks to families affected by this disorder, who participated in the research, the team was able to trace it back to its origin in the Salt Lake Valley nearly two centuries ago.
They leveraged advanced genome sequencing techniques and analysis of close to 6,500 datasets, ultimately identifying a segment of the ZFHX3 gene as SCA Type 4’s genetic root cause.
ZFHX3’s job in the body is to help recycle proteins. Part of ZFHX3 is much longer than it should be in people who develop SCA type 4, the research team found. Cells with this extended version of the gene show signs of being unhealthy — and failing to do this important protein recycling work.
“This mutation is a toxic expanded repeat, and we think that it actually jams up how a cell deals with unfolded or misfolded proteins,” neurologist Stefan Pulst from the University of Utah said in a news release.
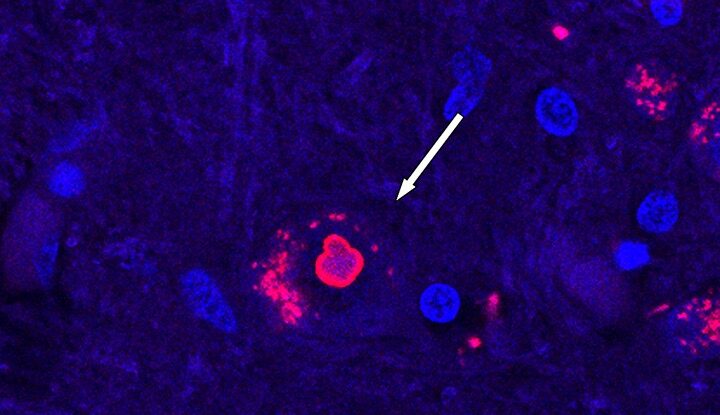
Pinpointing this gene has been difficult to date, in part because it’s so rare, and the findings wouldn’t have been possible without the involvement of patients and their families in the research. The breakthrough could pave the way for potential treatments and early detection, the team says.
“I’ve been working on SCA4 directly since 2010 when the first family approached me, and once you go to their homes and get to know them, they’re no longer the number on the DNA vial,” University of Utah neurologist Karla Figueroa said. “These are people you see every day… you can’t walk away. This is not just science. This is somebody’s life.”



This is incredible information about SCA .
We as a family, have been looking for an answer to our generational problems of unbalance, and gait issues.
Is double vision part of this rare disease?
Thank you so Much for your research and interest.
Wendy
Thank you for sharing your family’s experience, Wendy. Double vision may be a symptom associated with spinocerebellar ataxia (SCA), although the specific symptoms can vary depending on the type and individual. Take care.
Thank you Tori,
We are learning a little bit more about this disease, so I will keep in touch.
Grateful,
Wendy Richardson